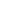苄亚甲基结构(Ar−CH2−R)构建简单,是合成化学中常见的结构单元,苄亚甲基基团修饰的分子在许多(>100种)药物中得到了应用。但由于其C−H键键能(90 kcal/mol)较弱,很容易发生代谢氧化,由此降低了药物的生物半衰期。双苄亚甲基(Ar−CH2−Ar)基团(C−H键键能82 kcal/mol)也不稳定,仅存在于17种已批准的药物中。如果用代谢稳定和亲脂性较强的苄二氟亚甲基(Ar−CF2−R)取代−CH2−基团可优化构效关系(SAR),但目前仍未得到有效的利用(图1a)。受限于这类结构基元的合成方法,目前仅有三种包含单苄二氟亚甲基(Ar−CF2−R)单元和一种双苄二氟亚甲基(Ar−CF2−Ar)的药物分子。
二氟甲基(R−CF2H)是药物化学中一类非常重要的结构基元,可以赋予药物分子良好的药代动力学特性,通常作为靶向的末端官能团,不会进一步修饰衍生化。对起始原料Ar−CF2H进行去质子化可以得到亲核性的Ar-CF2-合成子,这是一类尚未发掘但有望用于构建苄基Ar-CF2-R键的方法。近日,美国密西根大学(University of Michigan)化学系的Nathaniel K. Szymczak教授课题组在J. Am. Chem. Soc. 上发表文章,报道了在布朗斯特超强碱(Brønsted superbase)与弱路易斯酸的作用下,Ar-CF2H发生去质子化并对高反应性的Ar-CF2-进行捕获,提供了一种制备可分离、具有高活性Ar-CF2-合成子的方法。Ar-CF2-可作为亲核试剂在室温下与各种亲电试剂反应,不同二氟甲基(杂)芳香烃中均具有良好的适用性。
图1. (a)含有Ar-CF2-R基团的代表性药物分子;(b)Ar-CF2-R键的构建策略;(c)ArCF2H作为ArCF2-前体;(d)化合物1的合成。图片来源:J. Am. Chem. Soc.
现有的构建Ar−CF2−R键的方法需要使用有毒或者具有爆炸性的试剂,底物范围也很有限(图1b)。最常见的构建方法是断裂CF2的C−F键,通过酮羰基的去氧氟化或者C−H键的自由基氟化反应来实现。然而,每一种方法都具有一定的挑战性,酮的脱氧氟化会产生爆炸性的副产物,而C−H键的自由基氟化反应官能团兼容性较差。
与上述后期氟化策略相反,[ArCF2]结构单元参与的偶联反应能够实现更为模块化的合成。然而,亲电性的ArCF2+和ArCF2•自由基均存在显著的局限性:亲电试剂ArCF2Br起始材料来源有限,砜(R-CF2-SO2R)亲电试剂和芳基锌亲核试剂之间的自由基交叉偶联也仅限于脂肪族RCF2,从Ar-CF3形成Ar-CF2•则需要使用强还原剂,并且底物范围有限。ArCF2-亲核试剂参与的反应尚未得到有效的发展。ArCF2SiMe3试剂是该类反应唯一可用的试剂,但这类试剂的合成方法又非常有限。
二氟甲基芳香烃(ArCF2H)在去质子化后可能形成理想的掩蔽ArCF2-亲核试剂。基于过渡金属催化的二氟甲基化反应,这类化合物易于制备,并且能够实现结构多样化。然而,ArCF2H的C-H键酸性较弱,与KN(SiMe3)2不反应,如果发生去质子化,得到的ArCF2-片段也十分不稳定。选择能够与ArCF2H兼容的强碱和稳定氟烷基阴离子的路易斯酸有望解决这一问题。ArCF2H去质子化后形成的ArCF2-被路易斯酸捕获,随后与亲电试剂反应,释放ArCF2-。
使用布朗斯特碱/路易斯酸将ArCF2H基团转化为ArCF2-合成子必须满足三个关键要求:(1)用于C-H键去质子化的碱碱性必须足够强;(2)路易斯酸不能不可逆地结合布朗斯特碱(图1c);(3)路易斯酸同时具有ArCF2-捕获/稳定的能力,保证ArCF2-可以释放以促进随后与亲电试剂的反应。根据以往的报道,作者认为强碱PhCH2-和六甲基硼嗪(B3N3Me6,一种用于稳定氟烷基阴离子的路易斯酸)可以满足这些标准。他们在室温下将PhCF2H加入B3N3Me6、KCH2Ph和18-冠-6等摩尔混合的THF溶液中,以98%的产率和72%的分离产率(> 9 g规模)得到[K(18-冠-6)] [PhCF2-B3N3Me6](1)(图1d)。使用KC(Me)Ph2(3%)和KCHPh2(0%)参与反应产率显著降低。作者还考察了大位阻的二异丙基酰胺作为碱(Li、Na和KN(i-Pr)2),发现使用KN(i-Pr)2能以高产率(91%)得到产物,而NaN(i-Pr)2(21%)和LiN(i-Pr)2(0%)的效率较低。
作者接着考察了1将CF2Ph-转移到亲电试剂的能力。1可顺利对多种简单的含羰基和亚胺基团的亲电试剂(2a-2d,52-84%)进行苄二氟亚甲基化(图2),这些反应能够在室温下12小时内完成。与类似的CF3-转移试剂[K(18-冠-6)] [CF3-B3N3Me6]不同,1可以将PhCF2-转移至可烯醇化的羰基化合物(2e-2f,产率47%至62%)。叔胺、甲苯磺酰基、酯和芳基卤化物等都可以兼容。二氧化硫也可以经过1,2-加成得到二氟苄基硫酸钾(2i,49%),该产物是一种氧化活化的PhCF2•前体自由基,可对α/β不饱和对甲苯磺酰胺加成。除了1,2-加成反应,1也可以对二苯基二硫化物(Ph2S2)和溴(Br2)(2h,78%; 2g,85%)进行苄二氟亚甲基化。
图2. (a)1与酮、醛、亚胺、二硫化物、Br2、SO2及1,4-二硝基苯的反应;(b)吡啶C-H键的二氟苄基化;(c)Cr(0)(CO)3介导苯的C-H键二氟苄基化。(d)Pd介导的二氟苄基化。图片来源:J. Am. Chem. Soc.
使用以往的方法很难构建双苄基二氟亚甲基结构,唯一可用的方法是使用ArCF2Br作为原料,但该化合物必须通过自由基溴化或脱氧氟化策略来合成。而使用化合物1作为苄二氟亚甲基化前体,二硝基苯可以通过芳香亲核取代(SNAr)反应(2j,53%)直接进行苄二氟亚甲基化。当使用位阻大且活性强的路易斯酸B(C6F5)3活化时,吡啶也能与1反应生成路易斯酸稳定的阴离子σ-加合物,随后通过DDQ氧化、NMe4F消除保护基得到4-苄二氟亚甲基吡啶产物。这些反应可以通过一锅法在单一溶剂体系中进行。非杂环芳香烃(例如苯)可使用Cr(0)(CO)3进行活化,与1混合后再通过苯醌氧化得到稳定的σ-加合物,最终形成二苯基二氟甲烷(2n,72%)。
除了对活化的芳香烃进行苄二氟亚甲基化之外,作者还尝试了Pd介导的交叉偶联反应,1与廉价易得的芳基溴化物和碘化物混合,得到Ar-CF2-Ar。这是首次使用亲核性的ArCF2-作为转金属化试剂参与交叉偶联反应,为那些弱亲电性的芳基卤化物偶联体参与反应提供了一种互补策略。
图3. (a)ArCF2H去质子化作为亲核试剂的底物适用性考察;(b)将四种不同的ArCF2-亲核试剂和不同的亲电试剂混合。图片来源:J. Am. Chem. Soc.
接下来作者探究了其他二氟甲基芳香烃的活化情况。多种二氟甲基(杂)芳香烃在-80 ℃下与KN(i-Pr)2/B3N3Me6/18-冠-6的THF溶液混合,能以高产率形成稳定的ArCF2-合成子3a-3k(图3a)。当使用KCH2Ph代替KN(i-Pr)2时,产率显著降低。芳基溴化物不能兼容电化学和光氧化还原苄基氟化过程,易发生邻位金属化/苯炔化。然而,作者通过发展的方法可得到以上亲核ArCF2-物种。反应中不存在B3N3Me6时无法得到所需的产物由此说明了路易斯酸/强碱对ArCF2-捕获和转移的过程是协同的。
作者还将四种结构不同的二氟甲基(杂)芳香烃分别与四种有机亲电试剂(图3b,亚胺、醛、二硫化物和路易斯酸活化的吡啶)混合,通过两步反应得到16种产物(5a- 5p,41-87%)。其中一些产物含有独特的Ar-CF2-R结构,如咪唑-CF2-SR(5c)和噻吩-CF2-氨基甲酰胺(5d),这些结构可用于构建具有生物活性的非氟化分子。
总结
Nathaniel K. Szymczak教授课题组发展了一种将常见的二氟甲基(杂)芳香烃转化为稳定、结构多样的ArCF2-亲核试剂的方法,这在之前通过常规方法难以实现。得到的ArCF2-试剂具有高反应性,可与各种有机亲电试剂反应形成ArCF2-C键,由此可构建Ar-CF2-Ar结构。作者期待这种方法可以在药物分子合成的后期得到广泛的应用。
原文(扫描或长按二维码,识别后直达原文页面):
The Difluoromethyl Group as a Masked Nucleophile: A Lewis Acid/Base Approach
J. Am. Chem. Soc., 2018, 140, 9404, DOI: 10.1021/jacs.8b06093
导师介绍
Nathaniel K. Szymczak
http://www.x-mol.com/university/faculty/748
本文版权属于X-MOL(x-mol.com),未经许可谢绝转载!欢迎读者朋友们分享到朋友圈or微博!
长按下图识别图中二维码,轻松关注我们!
点击“阅读原文”,查看 化学 • 材料 领域所有收录期刊